Breakthrough in Atomic-Scale Mechanics: Scientists Conquer a Major Challenge in Material Simulation!
2024-12-09
Author: Emily
Introduction
In a groundbreaking achievement, a team of researchers has finally addressed one of the most daunting challenges in the computational mechanics of materials—achieving accurate simulations at an atomic scale.
The Challenge of Dynamic Simulations
Traditional computational methods, primarily relying on molecular dynamics (MD) with atomistic force-fields, have successfully described the stable phases of materials with complex chemical compositions. However, the real hurdle has been simulating how these materials respond mechanically to dynamic external stimuli.
The complexity arises from the very short time scales—mere femtoseconds—that MD methods operate on when attempting to model the precise motion and interactions of atoms. This limitation prevents effective simulation of the longer time scales seen in real-world experiments, like those observing deformation rates below 10 to 100 gigahertz. Essentially, this time-scale bridging issue has obstructed accurate computational predictions of material behavior in conditions typically ruled in standard mechanical tests and rheology.
Innovative Computational Framework
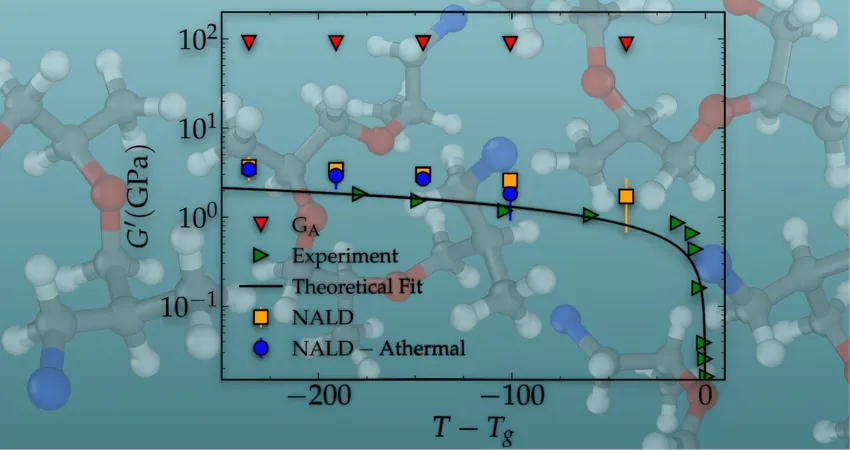
In a collaborative effort spearheaded by post-doc Dr. Vinay Vaibhav and Dr. Tim Sirk from the US Army Research Lab, this research team has unveiled an innovative computational framework that sets a new standard in molecular simulations. Their approach focuses on a concept known as nonaffine displacements, wherein atoms deviate from their prescribed trajectories during external deformation, creating what can be likened to Epicurus's philosophical idea of “clinamen.”
Understanding these nonaffine displacements has proven crucial, as they play a key role in maintaining mechanical equilibrium at every stage of deformation. This discovery has led researchers to compute vibrational normal modes, a method refined using the latest computational techniques, offering an unprecedented ability to simulate material behavior more accurately.
Significant Findings
In their published work in the esteemed journal *Macromolecules*, the team notably achieved a parameter-free alignment with the viscoelastic moduli of a real-world complex material—a crosslinked epoxy polymer glass in its amorphous state—at frequencies nearly 10 orders of magnitude lower than those observable using standard MD simulations. The striking correlation with experimental mechanical test data—without any adjustable parameters—highlights the robustness and reliability of this new computation method.
Future Implications
But that’s not all! The potential for future refinements is expansive. By utilizing larger material snapshots with a greater number of atoms, researchers anticipate enhanced predictive capabilities, which could reduce numerical fluctuations and noise.
The implications of this work are enormous. This method could illuminate the specific atomic and molecular vibrations responsible for the hardness or softness of various materials, paving the way for innovations in high-performance materials with diverse applications in technology and engineering. With this breakthrough, the future of material science looks not only promising but revolutionary!
Conclusion
Stay tuned for more updates on how this cutting-edge research evolves and reshapes the field!








 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)