Breakthrough Discovery Unveils Key Mechanism Behind Early Neurodegeneration in ALS
2024-11-08
Author: Yu
Introduction
In an alarming trend, around 5,000 people in the United States are diagnosed with amyotrophic lateral sclerosis (ALS) each year, with survival rates tragically hovering between two to five years post-diagnosis, as reported by the Centers for Disease Control and Prevention (CDC). This rapidly progressing neurodegenerative disease wreaks havoc on motor neurons in the brain and spinal cord, leading to muscle weakness, respiratory failure, and cognitive decline.
A Breakthrough Study
Despite the devastating implications of ALS, the initial triggers that set off the decline of motor neurons have remained elusive—until now. A new study from researchers at the University of California, San Diego, published in Neuron on October 31, 2024, illuminates a critical pathway that ignites neurodegeneration in the early stages of ALS, opening doors for potential therapies to halt or slow the relentless progression of this life-altering condition.
Role of TDP-43
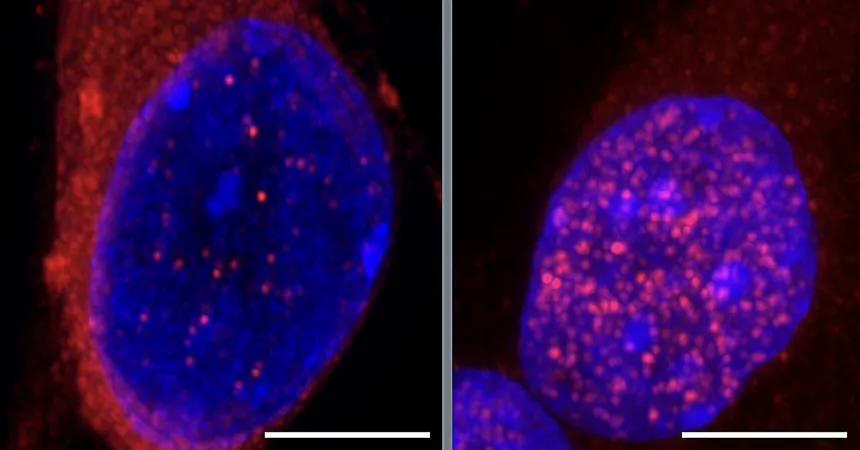
At the core of this discovery lies a protein known as TDP-43. Under normal circumstances, TDP-43 resides in the nucleus of motor neurons, where it plays a pivotal role in regulating gene expression essential for neuronal function. Alarmingly, when TDP-43 accumulates in the cytoplasm—an abnormal location for the protein—it signals a key indicator of ALS. Understanding how TDP-43 mislocalizes has perplexed researchers for years.
Cascade Triggered by CHMP7
As Gene Yeo, Ph.D., a lead researcher and professor at UC San Diego, poignantly stated, 'By the time you see a patient with ALS and see the TDP-43 protein aggregated in the cytoplasm, it's like the accident site with all the cars crashed already, but that's not the initiating event.' Delving deeper, Yeo and his team discovered that the misplacement of another protein, CHMP7, triggers a cascade of degenerative events in motor neurons.
Understanding the Mechanism
CHMP7, usually found in the cytoplasm, builds up in the nucleus, leading to damaging repercussions. But what sets off this nuclear accumulation? The researchers identified 55 RNA-binding proteins with potential links to ALS and, notably, when they inhibited the production of several of these proteins, CHMP7 levels surged within the nucleus.
One particularly intriguing finding involved the RNA-splicing protein SmD1. When researchers depleted SmD1—previously unlinked to CHMP7—surprisingly, they observed a significant increase in CHMP7 levels in the nucleus. This excess disrupts nucleoporins, tiny gateways that facilitate the transport of proteins and RNA between the nucleus and cytoplasm. A failure in nucleoporins allows TDP-43 to escape the nucleus, leading to further dysfunction of essential neuronal gene programs.
Potential Therapy Insights
Encouragingly, when the expression of SmD1 was enhanced, CHMP7 was restored back to its cytoplasmic locale, preserving the integrity of nucleoporins and allowing TDP-43 to remain in the nucleus. This pivotal adjustment spares motor neurons from degeneration—a promising revelation that points to potential therapeutic interventions.
Connecting ALS and SMA
Yeo's team also discovered SmD1's relationship with the SMN (survival motor neuron) complex, a crucial protein ensemble whose dysfunction is associated with spinal muscular atrophy (SMA), another severe neurodegenerative disorder. Current SMA therapeutics, such as risdiplam, are designed to enhance SMN2 gene splicing and expression. Yeo suggests a compelling possibility: could risdiplam be utilized not just for SMA, but to intervene in early ALS development?
Conclusion and Future Directions
“It's not like all the neurons die at once,” Yeo emphasizes. “Some neurons die first, and then there is spread across other neurons. Perhaps as soon as you get symptoms, we could treat the patient so that the rest of the neurons don't crash and hope to stop the progression of ALS.”
This significant breakthrough sheds light on the underlying mechanisms of ALS, but researchers acknowledge that much work lies ahead. The next steps include securing funding for research involving animal models and various genetic forms of ALS, with hopes of testing the efficacy of risdiplam and other compounds as potential halting forces against ALS. As the battle against this relentless disease continues, this discovery provides a glimmer of hope for earlier intervention and improved outcomes for individuals diagnosed with ALS.



 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)