Breakthrough in Atomic-Scale Mechanics: Scientists Tackle One of Materials Science's Toughest Challenges
2024-12-09
Author: Li
Groundbreaking Development in Material Mechanics
In a groundbreaking development, researchers are revolutionizing our understanding of material mechanics at the atomic level. Traditional computational methods used to simulate material properties are firmly rooted in molecular dynamics (MD) and rely on atomistic force fields. While these approaches excel at describing thermodynamically stable phases of complex materials, they have fallen short when it comes to modeling the mechanical deformation behavior of these materials in the face of external stimuli over extended time frames.
The Challenge of Time Scales
The crux of the issue lies in the exceptionally short time scales—mere femtoseconds—needed for molecular dynamics to function effectively. This limitation hampers the ability to simulate the dynamic response of materials during mechanical tests, especially for deformation rates that fall below the range of 10 to 100 gigahertz. This time-scale bridging problem has long stymied the accurate computational prediction of material performance in scenarios aligned with standard mechanical testing.
A Collaborative Breakthrough
However, a dedicated collaboration between Dr. Vinay Vaibhav, a post-doctorate researcher, and Dr. Tim Sirk at the US Army Research Laboratory, has opened new doors. Together, they have developed an innovative computational framework that addresses this critical challenge, marking an unprecedented advancement in molecular simulations of materials under stress.
The Role of Nonaffine Displacements
The team's groundbreaking approach pivots on a keen understanding of nonaffine displacements—atomic movements that deviate from the expected trajectory imposed by external forces. These displacements are crucial in maintaining mechanical equilibrium during deformation. As neighboring atoms exert forces on one another, they necessitate slight deviations—or 'swerve'—in their paths to stabilize the system, akin to the philosophical concept of 'clinamen' described by Epicurus.
Innovative Computational Techniques
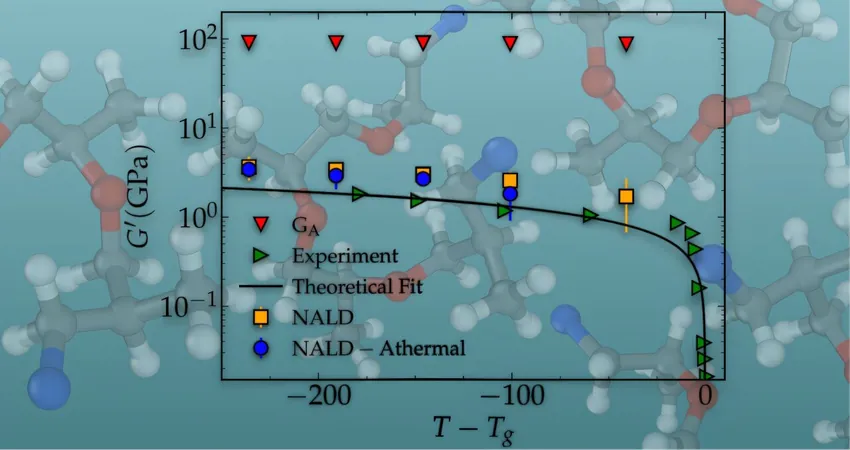
By implementing a mathematical model that harnesses these atomic movements, the researchers have employed modern computational techniques to compute the vibrational normal modes of materials successfully. This led to an impressive parameter-free agreement with the viscoelastic moduli of a complex crosslinked epoxy polymer glass in its amorphous state—achieved at frequencies a staggering ten orders of magnitude lower than traditional simulations could manage.
Unprecedented Results and Future Potential
The results are compelling, boasting a close match to experimental data without resorting to any adjustable parameters, which is a rare feat in material science research. Yet, the future holds even more potential for refinement, including enlarging the material configuration snapshots and incorporating more atoms to enhance predictions and minimize numerical fluctuations.
Implications for Material Science
Excitingly, this new framework could pave the way for identifying specific atomic and molecular vibrations central to a material's stiffness or softness. Such insights could catalyze the development of high-performance materials ideal for a myriad of applications in technology and engineering.
The Future of Materials Science
As scientists unravel the complexities of material behavior at such minute scales, the implications could herald a new era of innovative, resilient materials that push the boundaries of current capabilities in everything from electronics to aerospace engineering. Keep an eye on this transformative research as it unfolds—there's more to come that could change the landscape of materials science forever!

 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)