Kasuistik: Zwei Ohnmachtsanfälle an einem Tag - Ein tiefes Eintauchen in die Welt der kardialen Amyloidose
2024-11-26
Autor: Nina
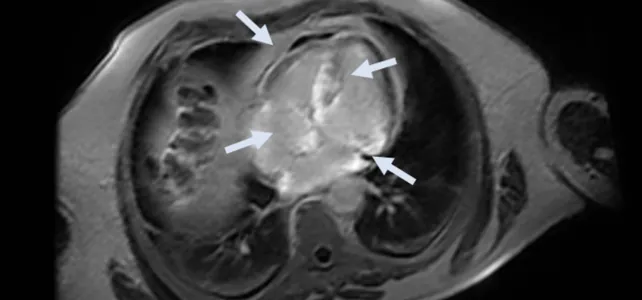
Im Rahmen von zwei akuten Synkopen wurden umfassende Untersuchungen durchgeführt, um die Ursache für die Ohnmachtsanfälle zu klären. Ein kardiales MRT (CMR) wurde als nächste diagnostische Maßnahme angeordnet. Die Ergebnisse zeigten eine biventrikuläre Hypertrophie mit erhaltener systolischer Funktion und ein infiltratives Muster mit ausgeprägten Bereichen von späten Gadolinium-Enhancements (LGE) in verschiedenen Bereichen des Herzens.
Das EKG ergab niedrige QRS-Spannungen, die typisch für eine kardiale Amyloidose sind, aber nur bei einem Drittel der Patienten mit ATTR-Amyloidose nachgewiesen werden. Weitere EKG-Veränderungen umfassten Anomalien in der Erregungsleitung und rhythmische Störungen. Das transthorakale Echokardiogramm (TTE) bestätigte eine konzentrische Hypertrophie mit diastolischer Dysfunktion und Klappenverdickung.
Angesichts dieser Befunde wurde der Verdacht auf eine kardiale Amyloidose erhärtet. Es wurde ein Ganzkörper-SPECT/CT zur Knochenszintigraphie durchgeführt, das eine starke Fixierung des Radiopharmakons in den septalen und lateralen Regionen des linken Ventrikels zeigte, mit einer Perugini-Score-Bewertung von 3, was auf eine signifikante myokardiale Ablagerung hinweist.
Eine elektrophysiologische Untersuchung ergab eine subhisianische Reizleitungsstörung, was zur Entscheidung führte, ein CRT-P (Cardiac Resynchronization Therapy Pacemaker) zu implantieren. Nach eingehender Analyse und molekularer Untersuchung des TTR-Gens wurde die Diagnose einer Transthyretin-bezogenen Wildtyp-Amyloidose (ATTR-wt) gestellt.
Die kardiale Amyloidose, besonders die ATTR-Form, ist eine restriktive Kardiomyopathie, die durch Ablagerungen von strukturell veränderten Proteinfragmenten im Herzgewebe gekennzeichnet ist. Es gibt zwei Hauptformen: die nicht erbliche (ATTR-wt) und die vererbbare Form (ATTRv), welche durch Mutationen im TTR-Gen verursacht werden. Die ATTR-wt-Form betrifft vor allem ältere Männer und kann zu spezifischen Symptomen wie Herzinsuffizienz, Dyspnoe und kardiovaskulären Komplikationen führen.
Besonders bemerkenswert sind die neurologischen Symptome, die mit ATTR-Amyloidose einhergehen können, einschließlich peripherer Neuropathien und autonomer Dysfunktionen. Das Karpaltunnelsyndrom, häufig bilateral, kann ein frühes Zeichen der Erkrankung sein und tritt oft Jahre vor den kardialen Symptomen auf.
Die Diagnose der kardialen Amyloidose kann herausfordernd sein, da die klinischen Symptome unspezifisch sind. Die kardiale Magnetresonanztomographie (CMR) hat sich jedoch als hervorragendes nicht-invasives Verfahren zur Beurteilung und Diagnose erwiesen. Der Einsatz von nuklearmedizinischen Verfahren hat die Diagnose weiter revolutioniert, da radioaktive Isotope spezifisch an Amyloidablagerungen binden.
Zusammenfassend lässt sich sagen, dass eine frühzeitige Diagnostik und Behandlung entscheidend für das Management der kardialen Amyloidose sind. Ohne adäquate Therapie kann die Erkrankung zu schweren kardiovaskulären Komplikationen führen, einschließlich Herzinsuffizienz und plötzlichen Herzereignissen.


 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)